7/20に塩野義の新型コロナウイルス経口治療薬ゾコーバ(一般名:エンシトレルビル)の審査が行われました。
Twitterでも色々と記載させて頂きましたが、結果的には継続審議となりましたね。
今日はそんなゾコーバの審査報告書を読んでみて、有効性や安全性について、重要だと思われる点について簡単に解説してみます。
前評判でも色々とありましたが、果たしてPMDAはどのように判断していたのでしょうか?
医薬品の審査に国産云々といったものは影響せず、「有効性と安全性を中心とした科学的な視点」で審査が進められていることをご理解頂けますと嬉しいです。
なお本記事は業界外の方でも理解できるように、多少表現を丸めている部分もあります。
詳しくは審査報告書をご覧ください。
無料で誰でも閲覧することができます。
あと結構大事なところが多くて、1万文字超えちゃいましたので、必要に応じて流しながら読んで頂けますと幸いです。
前編後編とか嫌いなので一気に書いちゃいました!
吹き出しだけ読んでいれば要点はつかめるようにしているつもりです。
Contents
概要
ゾコーバは塩野義の開発した新型コロナウイルス感染症に対する抗ウイルス薬です。
軽症から使える国産の飲み薬ということで、政治家等に持ち上げられたこともありましたね。
作用機序はウイルスのプロテアーゼの阻害によるウイルス複製の阻害です。
作用機序についてもう少し詳しく見てみましょう。
SARS-CoV-2が細胞に侵入すると、ウイルスRNAを鋳型にタンパク質が翻訳されます。
このうち、非構造タンパク質はポリタンパク質として翻訳され、ポリタンパク質がSARS-CoV-2 3CLプロテアーゼ等によるプロセシングを受け、ウイルスの複製等に必須である非構造タンパク質となります。
ゾコーバはこのSARS-CoV-2 3CLプロテアーゼに対する阻害活性を有し、ポリタンパク質のプロセシングを阻害することによりSARS-CoV-2の複製を阻害することで、ウイルスを減らすということです。

この「プロテアーゼ阻害」という作用機序については、概ねファイザーのパキロビッドと同じと考えてよいでしょう。
着眼点やアプローチは同じということです。
同じようにアプローチするということは有効性や安全性も似た傾向となるケースが多いですね。
あくまで傾向ですが。
申請データパッケージ
ゾコーバの臨床試験はⅡaやⅡb等、ややこしくもありますので、簡単に申請データパッケージをおさらいしておきましょう。
よくわからないという方は、ここは読み飛ばしていただいても構いません。
ゾコーバの有効性や安全性を確認する主な臨床試験として、国際共同第Ⅱ/Ⅲ相試験(T1221試験)があります。
この試験は4つのパートから構成され、第Ⅲ相パート及び第Ⅱb/Ⅲ相パートは試験実施中となります。
今回の申請については、第Ⅱa相パートと第Ⅱb相パートの成績に基づき、検討が行われています。
・第Ⅱa 相パート:結果あり
軽症~中等症のSARS-CoV-2 による感染症患者及び無症状のSARS-CoV-2 病原体保有者を対象として本薬の抗ウイルス活性を確認する
・第Ⅱb 相パート:結果あり
軽症~中等症のSARS-CoV-2 による感染症患者を対象として本薬の臨床症状の改善及び抗ウイルス活性を確認する
・第Ⅲ相パート:結果まだ
軽症~中等症のSARS-CoV-2 による感染症患者を対象として本薬の有効性を検証する
・第Ⅱb/Ⅲ相パート:結果まだ
無症状のSARS-CoV-2 病原体保有者及び軽度の症状のみを有するSARS-CoV-2 による感染症患者を対象として本薬の有効性を検証する
第Ⅱb 相パートの主要評価項目は、第Ⅱa 相パートの結果も参考に、
「SARS-CoV-2による感染症の12症状合計スコアの治験薬投与開始(Day 1)から120時間(Day 6)までの単位時間当たりの変化量及びDay 4におけるSARS-CoV-2のウイルス力価のベースラインからの変化量」のcoprimary endpoint と設定されています。
なおこのendpointについては試験実施中に決定されています。
これについて機構は「検証的試験の実施中に大きな計画変更を行ったことは適切ではなかったと考えるが、SARSCoV-2 による感染症の流行状況の変化を踏まえると、試験計画の変更について一定の理解はでき、また、二重盲検下での変更であったことから、当該変更後の結果を評価することは可能と考える」としています。
なおendpoint設定についてですが、臨床症状の評価項目においては、下記の事項を踏まえ、12 症状合計スコアの各時点のベースラインからの変化量について治験薬投与開始(Day 1)から120 時間(Day 6)までのAUC を台形法で計算し、AUC 算出時の評価期間(単位:時間)で割った値で評価することとしています。
・治験薬投与終了翌日のDay 6 時点におけるベースラインからの変化量で評価した場合、Day 6 以前の治療効果を表す変化量の情報を間接的にしか考慮できないこと
・他のSARS-CoV-2 による感染症の治療薬において症状合計スコアの変化量のAUC を副次評価項目にしていること
・各症状の累積が患者にとっての臨床上の負担であり、その負担を軽減することに臨床的意義があると考えたこと
・12症状合計スコアが早期に低下することが症状回復までの時間を短縮することに寄与すると考えたこと
これについては機構に意義は不明と言われてしまっていますね。
ちょっとかわいそう。
また第Ⅱa 相パートのプラセボ群のうち、SARS-CoV-2 による感染症の症状を有する被験者において、Day 6 時点で多くの被験者でウイルス力価が検出限界未満であったこと等を踏まえ、ウイルス力価の評価時点はDay 4と設定しています。
また、ウイルスRNA を測定する場合、感染性を失ったウイルスのゲノム断片まで捉えてしまうことから、感染力を有するウイルスを定量的に測定するウイルス力価の方が臨床的意義が高いと考え、ウイルスRNA ではなくウイルス力価を指標としています。
加えて変化量を指標とすることで、ウイルス力価の検出有無よりも抗ウイルス効果を定量的に感度よく評価可能と考えられています。
上記を踏まえ、本剤の有効性評価として、抗ウイルス効果を示し、それが臨床的な効果に反映されていることを示せるように、薬理的エンドポイントと臨床的エンドポイントの両者を主要評価項目として設定したとのことです。
抗ウイルス効果だけで有効性を示すことは受け入れられないだろうという読みが見て取れます。
これは過去に承認されたゾフルーザのその後の評判や、パキロビッド等の既存のCOVID-19治療薬の主要評価項目や結果も踏まえて設定したのかなと推測しています。
有効性
では大事な有効性についてみていきましょう。
この項目では国際共同第Ⅱ/Ⅲ相試験の第Ⅱb 相パート<2022 年1 月~3 月>を中心に見ていきます。
この試験はSARS-CoV-2 による感染症患者[目標例数435 例(各群145 例)]を対象に、有効性及び安全性を検討することを目的として実施された、プラセボ対照無作為化二重盲検並行群間比較試験です。
日本及び韓国の2カ国88施設で実施されています。
用法・用量は少々複雑でして、下記のようになっています。
①1 日目は本薬375 mg を1 日1 回、2 日目から5 日目は本薬125 mg を1 日1 回経口投与(以下、「本薬375/125 mg 群」)
②1 日目は本薬750 mg を1 日1 回、2 日目から5 日目は本薬250 mg を1 日1 回経口投与(以下、「本薬750/250 mg 群」)、
③プラセボを1 日1 回5 日間経口投与
主要評価項目は前述のとおり、
・SARS-CoV-2 による感染症の12 症状合計スコアの治験薬投与開始(Day 1)から120 時間(Day 6)までの単位時間当たりの変化量
・Day 4 におけるSARS-CoV-2 のウイルス力価のベースラインからの変化量
上記2つのco-primary endpoint となっています。
結果を下記に引用します。
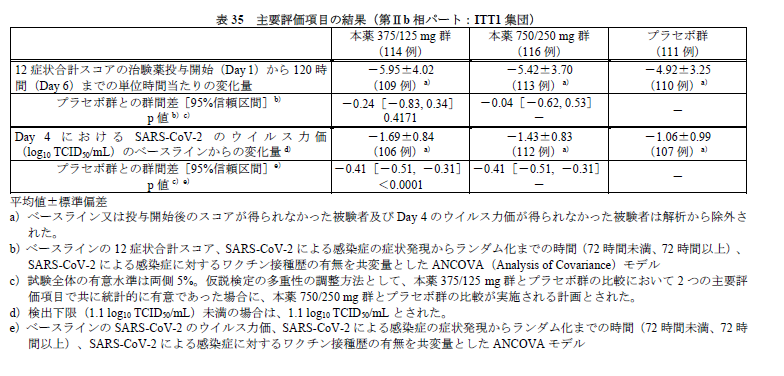
それぞれの項目について見ていきましょう。
主要評価項目
ウイルス力価について、本薬375/125 mg 群とプラセボ群、本薬750/250 mg 群とプラセボ群の間で統計学的な有意差は認められました。
しかし仮説検定の多重性は調整されていません。
とはいえ、ウイルス力価の減少は大きく、その点の効果は十分であると見て取れます。
(そこに臨床的意義があるかは別のお話)
ただ用量依存性が見られておらず、なんだかよくわからない結果という印象もあります。
また後述のもう一つのendpointである臨床症状の部分で主要評価項目を満たしておらず、多重性の調整もなされていないことから、この結果を大っぴらに語るのはやや問題があると思います。
では次に臨床症状の効果を見てみましょう。
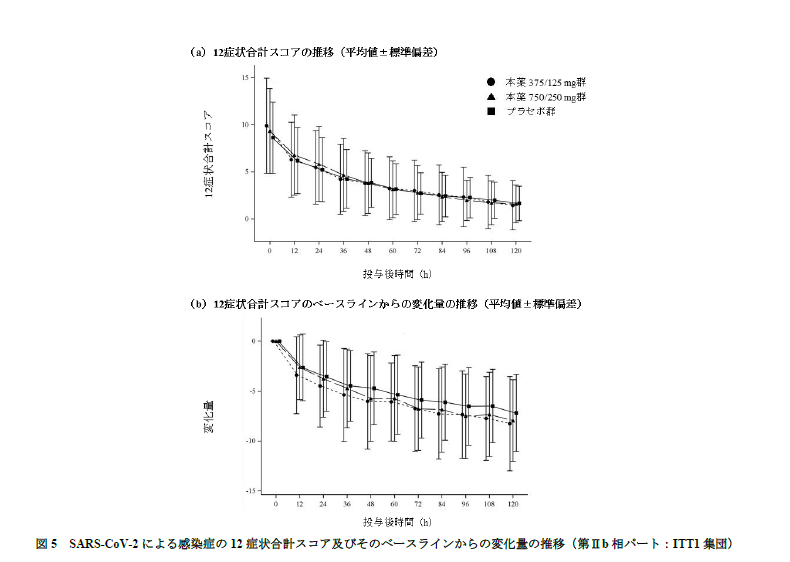
SARS-CoV-2 による感染症の12 症状合計スコアの変化量について、プラセボ群と比較していずれの本薬群でも統計学的な有意差は認められず、主要評価項目は未達でした。
図5を見ると、これは全然だめだなぁというのが見て取れますね。
事後解析:オミクロン株に特有な症状への効果
これではお話にならないということで、塩野義としてはオミクロン株に対する効果を示しています。
オミクロン株に特徴的な呼吸器症状[鼻水又は鼻づまり、喉の痛み、咳、息切れ(呼吸困難)]のに対する結果は表38 のとおりです。
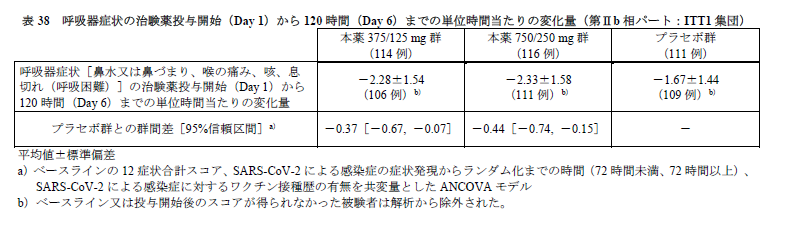
仮説検定の多重性は調整されていないものの、これらの症状においては、プラセボ群と比較して、本薬375/125 mg 群及び本薬750/250 mg 群のいずれにおいても統計学的に有意な差が認められております(それぞれ両側p 値はp=0.0153、p=0.0033)。
また、オミクロン株に特徴的な症状に対する臨床症状改善効果を確認するために、事後解析として、SARSCoV-2 による感染症の12 症状のうち、ベースラインにおけるスコアの平均値が1 以上であった症状(鼻水又は鼻づまり、喉の痛み、咳、熱っぽさ又は発熱)に、SARS-CoV-2 による感染症の重症度分類の指標の一つである息切れ(呼吸困難)を加えた5 症状について、治験薬投与開始(Day 1)から120 時間(Day 6)までの単位時間当たりの変化量(平均値±標準偏差)を確認したところ、下記のような結果でした・
・本薬375/125 mg 群で-3.17±1.79
・本薬750/250 mg 群で-3.26±1.81
・プラセボ群で-2.49±1.66
仮説検定の多重性は調整されていないもののプラセボ群と比較していずれの本薬群においても統計学的に有意な差が認められています(それぞれ両側p 値はp=0.0164、p=0.0039)。
上記の結果を総合的に勘案すると、本剤のSARS-CoV-2 に対する抗ウイルス効果及び臨床症状改善効果が確認されたと「塩野義」は考えていました。

これに関して機構の見解をもとに考えていきましょう。
この第Ⅱb 相パートでは2つの主要評価項目をco-primary endpoint と位置付けており、2つの主要評価項目の間で仮説検定の多重性の調整は計画されていませんでした。
多重性、多重性と一体何だと思われる方もおられるかもしれませんが、
検定を複数回実施し、かつ意思決定の選択肢が複数存在すると、本来は「効果がない」被験薬であっても、誤って「効果がある」と判断してしまう確率が有意水準を超過することが知られています。
これを検定に伴う多重性の問題といいます。
複数の主要評価項目が設定される場合は、複数の検定仮説が存在することから、
検定の順序や意思決定の枠組み(1 つでも有意な結果があれば試験成功とするのか、複数の項目で有意な結果が得られないと試験成功と見なさないかなど)を予め考慮しておく必要があるのです。
しかしこの試験ではそれが行われていなかった。。。
まぁ御託はおいておいて、結論をまとめると
「12症状合計スコアの治験薬投与開始(Day 1)から120 時間(Day 6)までの単位時間当たりの変化量」について統計学的な有意差が認められていないため、「Day 4 におけるSARS-CoV-2 のウイルス力価のベースラインからの変化量」についての統計学的な有意差についての評価はできないということです。
また機構としては、本薬による臨床症状の改善効果について、第Ⅱb 相パートの主要評価項目の一つとして「SARS-CoV-2 による感染症の12 症状合計スコアの単位時間当たりの変化量)(AUC を120 時間で割った値)」が設定されているが、最終評価時点の転帰と評価が一致しない可能性もあり、症状スコアの推移をAUC により評価することの意義は不明であるとしています。
そして12 症状合計スコアの推移はプラセボ群で概ね同様でしたね。
塩野義としては事後解析で、12 症状のうち、オミクロン株に特徴的な症状と考えられる「呼吸器症状[鼻水又は鼻づまり、喉の痛み、咳、息切れ(呼吸困難)]の合計スコア」及び「ベースラインにおけるスコアの平均値が1 以上であった症状(鼻水又は鼻づまり、喉の痛み、咳、熱っぽさ又は発熱)に、重症度分類の指標の一つである息切れ(呼吸困難)を加えた5 症状の合計スコア」は単位時間当たりの変化量がプラセボ群と比較して本薬群で大きかったと説明していますが、機構は下記の事項から臨床症状の改善効果は確認できたとは言えないとバッサリ切っています。
・多様な症状を呈する SARS-CoV-2 による感染症において、一部の症状スコアの結果から臨床症状の改善効果を解釈することには限界があること
・単位時間当たりの変化量の比較ではプラセボ群と比較して本薬群の変化量が大きい傾向が認められているが、群間差の推定値は各症状スコアの最小単位である1 を下回り、これらの症状について意義のある群間差が認められているとは解釈できないこと
・臨床症状に係るいずれの副次評価項目についても、プラセボ群と本薬群の結果に明らかな相違は認められていないこと
臨床試験において、プラセボ群と比較して本薬群でウイルスRNA 量やウイルス力価が低下することを確認することは重要であると考えるものの、 SARS-CoV-2 による感染症は、通常は、自然経過においても比較的短期間でウイルス量が減少することから、ウイルス力価の減少の臨床的意義を評価することは困難であること
雑にまとめると、
安全性
次に安全性についての試験結果を見てみましょう。
主に臨床試験の結果についてみていきますので、非臨床試験について詳しくは述べませんが、本剤の承認審査において、大きく注目された催奇形性と薬物相互作用の部分は見ておく必要があると思いますので、その部分は簡単にまとめてみました。
催奇形性
ラットを用いた受胎能及び着床までの初期胚発生に関する試験、ラット及びウサギを用いた胚・胎児発生に関する試験、ラットを用いた出生前及び出生後の発生並びに母体の機能に関する試験が実施されています。
その結果、主な毒性所見として、ラットにおいて胎児の骨格変異及びウサギにおいて胚・胎児死亡、胎児の軸骨格に奇形・変異、外表に奇形所見として短尾及び二分脊椎が認められています。
これについて機構は、ウサギを用いた胚・胎児発生に関する試験において、胎児の軸骨格及び外表に奇形を示唆する所見が認められたことから、潜在的な催奇形性リスクを有するとしています。
ウサギ胚・胎児において上記の毒性所見に対する本薬の無毒性量を投与した場合と、ヒトにおける本剤投与時の本薬の血漿中曝露量との曝露比は約2.4倍であり、十分な安全域を有していません
2.4倍というのはかなり低いですね。
また、臨床試験の除外基準として、当然ではありますが、妊娠している可能性のある女性及び妊婦が設定されていたことから、妊婦への投与に関する安全性情報は得られておらず、ヒト胎児に対する安全性は確立していません。
また、日本において、SARS-CoV-2 による感染症に対する治療薬で妊婦に対して投与可能な薬剤も承認されていることを考慮すると、ゾコーバを妊婦に対して使用するなんてことはせず、妊婦又は妊娠している可能性のある女性に対する本剤の投与は禁忌に設定することが適切であるとされています。
妊娠可能性がなければいいじゃん。高齢者に使えばいいじゃん!と言われる方もおられますが、野放しにして全て適切に使用されるというのは、とんでもない性善説です。
催奇形性については過去にサリドマイドによる極めて重大な薬害が発生しており、当局としてもより慎重な姿勢で臨むのは当然のことだと思います。
参考

薬物相互作用
in vitroの検討から、本薬は主にCYP3A で代謝され、P-gp 及びBCRP の基質であることが示唆されています。
併用薬がゾコーバに与える影響
まずは併用薬がゾコーバに与える影響についてみてみましょう。
といってもこの部分はかなり専門的な解説となってしまいますので、塩野義/当局の一致した見解だけ記載しておきます。
・CYP3A阻害薬との併用について、臨床的に問題となる薬物相互作用を示す可能性は低く、添付文書における注意喚起は不要
・ヒトにおける本薬のBA は高いことが想定され、消化管での吸収過程において本薬はP-gp 又はBCRP の阻害の影響を受けにくいと考えられる
・本薬とCYP3A 誘導薬を併用することで本薬の血漿中濃度が低下し、本薬の有効性に懸念が生じる可能性があることから、影響の大きさを勘案し、強いCYP3A 誘導薬は併用禁忌、中程度のCYP3A 誘導薬は併用注意として添付文書において注意喚起を行う
ゾコーバを代謝する酵素を強くする薬を一緒に使うと、ゾコーバが消えるのが早くなり、ちゃんとした効果がでなくなっちゃうよー!ということです。
ゾコーバが併用薬に与える影響
次にゾコーバが併用薬に与える影響を見てみましょう。
ここも色々とあるのですが、結論から言うと塩野義の検討したPBPK解析モデルには問題があり、もう一回やりなおせよ!と言われており、塩野義としても薬物相互作用の試験をやり直すとのことです。
現行で得られている結果としては、現時点では、CYP3A4阻害剤として併用禁忌及び併用注意を設定し、やりなおした試験結果が得られた後に再検討することとしたいとされています。
簡単に言うと、ゾコーバが薬を分解する酵素の働きを弱めてしまうため、その代謝酵素で分解される医薬品が分解されなくなり、血中濃度が上昇して危ないから、一緒に使うのはまずいんじゃないの?ということですね。
耐性プロファイルについて
in vitroでの検討において、本薬存在下で、SARS-CoV-2 3CL プロテアーゼにアミノ酸変異(D48G、M49L、P52S、S144A 及びM49L/S144A)が認められ、これらのアミノ酸変異を有する場合、本薬に対する感受性が低下することが確認されています。
ではこれが臨床試験においてこれがどう影響しただろうか?ということですが、
国際共同第Ⅱ/Ⅲ相試験(T1221 試験)第Ⅱa相パートにおいてアミノ酸変異が認められた症例で、ウイルス力価及びウイルスRNA量の再上昇は認められていませんでした。
つまり現行の結果からは気にしなくてよいかも?というところです。
しかしPMDAとしては、アミノ酸変異と本薬の有効性との関連も含め、臨床試験における本薬に対する耐性変異に関する情報は極めて限られており、in vitro において本薬に対する感受性が低下した変異株が認められていることを踏まえると、本薬を臨床使用した場合に、耐性変異株が出現し、本薬の有効性に影響を及ぼす可能性は否定できないとしています。
そのため国際共同第Ⅱ/Ⅲ相試験(T1221 試験)第Ⅱb 相パートの解析結果が得られ次第、速やかに評価し、新たな知見が得られた場合には適切に対応する必要がある。
また耐性変異の発現の有無は本薬の有効性に関する重要な情報であることから、公表文献等も含めて引き続き収集し、新たな知見が得られた場合には、適切に対応する必要があるとしています。
広く臨床で使用されれば、ゾフルーザのように耐性プロファイルで大きな問題が発生する可能性もないとは言えませんね。
副作用について
次に主な副作用についてみてみましょう。
結論から申し上げますと、現状ではそこまで気にすべき事象はないと思いました。
ただし検証は不足しています。
Ⅱb試験における有害事象/副作用は、下記の通りでした。
・本薬375/125 mg 群で34.3%(48/140 例)/13.6%(19/140 例)
・本薬750/250 mg 群で42.9%(60/140 例)/22.1%(31/140 例)
・プラセボ群で31.2%(44/141 例)/5.0%(7/141 例)
この試験では死亡に至った有害事象は認められませんでした。
重篤な有害事象は、プラセボ群2 例(胸椎骨折及び顔面麻痺各1 例)に認められ、いずれも治験薬との因果関係は否定され、転帰は軽快/回復となっています。
重篤な有害事象及び死亡に至った有害事象は認められていない一方で、プラセボ群と比較して本薬群で有害事象及び副作用の発現割合が高い傾向が認められています。
有害事象/副作用の内訳の詳細は審査報告書をご覧いただければと思いますが、特に高比重リポ蛋白減少(HDL コレステロール減少)が本薬群で高頻度に認められております
また、本薬750/250 mg 群では、他の脂質関連事象の発現割合もプラセボ群より高い傾向が認められています。
現時点では、臨床上の大きな懸念となる可能性は低いと考えられていますが、プラセボ群と比較して高頻度に発現している高比重リポ蛋白減少について添付文書において注意喚起する必要があるとされています。
副作用の面では大きな問題はなかったといえるでしょう。
しかし後述しますが、投与例数が少ないため、安全性プロファイルについては現状では検証不足であると思います。
安全性のまとめ
催奇形性も非臨床試験で示唆されており、CYP3A4阻害作用から、併用薬との相互作用にも注意が必要である。そして耐性プロファイルの面でも、広く使われたら、もしかしたらゾフルーザのように耐性ウイルスで問題が発生するかもしれないという点が懸念事項かなと思います。
ファイザーのパキロビッドと直接比較をしているわけではありませんが、現行の結果から「安全性面において優位性がある。より使いやすい薬になりえるとは言い難い」でしょう。
この点において優位性が示せたのであれば、使える患者が広がるという面で社会的な意義も大きかったものと思われますが、これではちょっと厳しかったですね。
副作用という観点からは、第Ⅱa 相及び第Ⅱb 相パートの結果を踏まえると、安全性上の大きな懸念は認められず、一定の忍容性は示されていると思われますが、まだまだ例数が少なく、製造販売後に多くの患者に使用された場合に、新たな安全性上の懸念が生じる可能性は否定できないといえるでしょう。
そのため現段階ではまだまだ検証不足と思われます。
これについても大規模な検証試験を実施済みで、安全性上大きな問題が認められていないパキロビッドと比較して、優位性があるとは言い難いかと思われます。
むしろ情報が不足しているともとれる状態です。
臨床的な位置づけについて
ゾコーバの臨床的な位置づけについて、塩野義はどう考えているのか?それに対して機構はどのように考えたのか?をまとめてみましょう。
塩野義としては、
無症状の SARS-CoV-2 病原体保有者及び酸素投与を要しないSARS-CoV-2 による感染症患者を対象として実施した国際共同第Ⅱ/Ⅲ相試験(T1221 試験)の第Ⅱa 相パート及び第Ⅱb 相パートの結果から、
本薬のSARS-CoV-2 に対する抗ウイルス効果及び臨床症状改善効果が確認されており、安全性上の特段の懸念は認められていないことから、ウイルス感染後早期に本剤の投与を開始することで、ウイルス増殖を速やかに抑制し、ウイルス感染に起因する過剰な炎症や免疫反応を抑えて臨床症状を改善し、さらには、入院又は宿泊療養等による隔離期間を短縮することで、ウイルス感染による患者への負担及び医療資源の逼迫の軽減にもつながると考えています。
また、当該試験の第Ⅱa 相パート及び第Ⅱb 相パートにおいて、SARS-CoV-2 に対するワクチンを1 回以上接種済みであった被験者の割合は約8 割であったこと、第Ⅱb 相パートにおける、SARS-CoV-2 による感染症の重症化リスク因子を有する被験者の割合は約3 割であったことを踏まえると、本剤はワクチン接種の有無やSARS-CoV-2による感染症の重症化リスク因子の有無にかかわらず、無症状のSARS-CoV-2 病原体保有者及び酸素投与を要しないSARS-CoV-2 による感染症患者に対して広く使用される抗ウイルス薬として新たな治療選択肢となり得るとしています。
これに対する機構の見解としては、
国際共同第Ⅱ/Ⅲ相試験(T1221 試験)の第Ⅱb 相パートの成績に基づき、本剤の有効性が推定できるとは判断できず、現時点では、本剤がSARS-CoV-2 による感染症の治療選択肢になるとは判断できない
とぶったぎっています。また、
現時点で得られている情報に基づき、本剤が承認される場合には、効能・効果及び適用対象は、既承認の経口治療薬と同様に、SARS-CoV-2 による感染症及びSARS-CoV-2 による感染症の重症化リスク因子を有する等、治療薬の投与が必要と考えられる患者とし、他の治療薬が使用できない場合に限り本剤を使用することが妥当と考えるとしています。
これは非常に厳しい見解です。
ここまで突き放されては、もはやどうしようもないでしょう。
有効性の推定ができないだけではなく、臨床的な位置づけすら危うい状況です。
この時点で趨勢は決していたといっても過言ではありません。
機構にRejectされたようなものですので、よくこの内容で2回目の審議にこぎつけたなと思うのが正直なところです。
言ってしまえば超絶甘々対応です。
まとめ
さてそれでは審査報告書における有効性/安全性の評価をまとめてみましょう。
有効性について、ウイルス量が減少する傾向が認められていることは否定しないが、有効性が推定できるものとは判断できず、第Ⅲ相パートの結果等を踏まえて改めて検討する必要がある
安全性について、大きな懸念は認められず、一定の忍容性は示されていると考えるが、投与経験は限られており、本剤が製造販売後に多くの患者に使用された場合に、新たな安全性上の懸念が生じる可能性は否定できない。
なお、現時点で得られている情報等を踏まえて、本剤が承認される場合には、添付文書において、催奇形性リスク及び薬物相互作用を含めて適切に注意喚起を行う必要がある。
また、実施中の国際共同第Ⅱ/Ⅲ相試験(T1221 試験)の第Ⅱb/Ⅲ相及び第Ⅲ相パートの情報を含め、さらに安全性の検討を行い、新たな知見が得られた場合には適切に医療現場に情報提供する必要があると考える。
この内容も踏まえて20日の審議が行われたわけです。
今までの内容を読んでも塩野義の治療薬が忖度で承認されなかったと思われますか?
国産だからとひいきせず、科学的な観点で医薬品の審査が行われて、当たり前のことでありながら一安心と言ったところです。
塩野義さんとしてもくじけずに、順当に第Ⅲ相パートで結果を残して、秋にリベンジしていただきたいですね!